近日,OD网页版,OD(中国)官方韩永昌教授与大连化学物理研究所分子反应动力学国家重点实验室傅碧娜研究员及张东辉院士合作,在之前H + C2H4→H2+ C2H3中发现碰撞诱导漫游(collision-induced roaming)机理的基础上[Chemical Science, 11, 2148 (2020)],在另一个体系H + C2H2 →H2+ C2H中再次发现了新奇的漫游反应机理[Journal of Physical Chemistry Letters, 12, 4211 (2021)]。
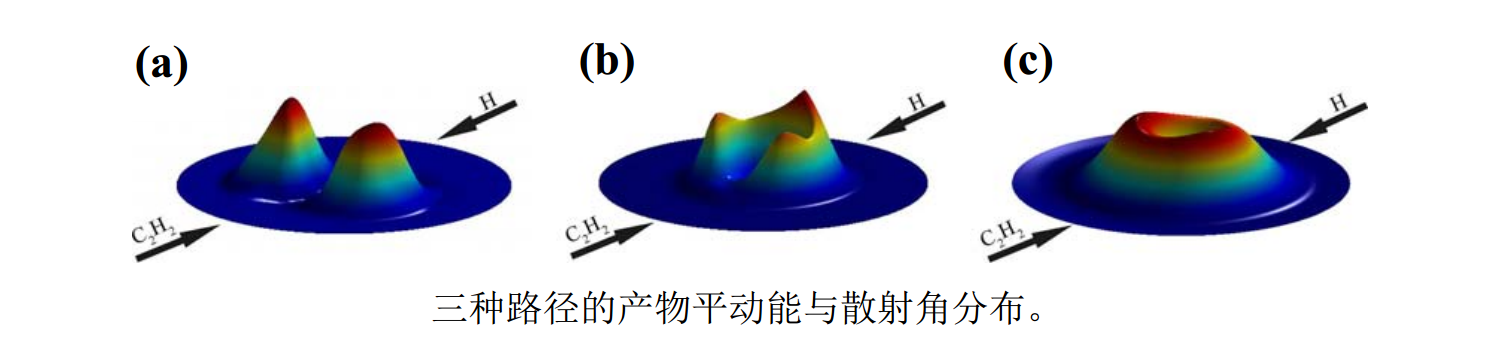
基于新构建的高精度全维势能面用准经典轨线方法开展了H + C2H2碰撞的动力学研究,发现除了常规的过渡态反应机理外,还存在漫游反应机理(反应路径中原子团结构发生巨大形变且不经过过渡态结构)。根据漫游过程中目标分子的结构,将漫游路径进一步分为A-漫游(Acetylene-facilitated roaming)和V-漫游(Vinylidene-facilitated roaming),分别对应漫游H原子的撞击目标乙炔分子和亚乙烯基分子。三种路径的产物能量与空间角分布都完全不同,而且在H + C2H2→H2+ C2H反应中漫游机理起主导作用。
相关研究成果发表在《The Journal of Physical Chemistry Letters》上,该期刊为原子与分子物理领域顶级期刊之一,影响因子为6.710,OD网页版,OD(中国)官方博士生付艳林为论文第一作者,韩永昌教授为共同通讯作者。
(a)传统路径:H + HCCH→[TS] (传统过渡态为5原子共面结构)→H2+C2H
(b)新路径A-漫游:H + HCCH→[H* … HCCH] (目标乙炔分子)→H2+C2H
(c)新路径V-漫游:H + HCCH→[H* … H2CC] (目标亚乙烯基分子)→H2+C2H
文章链接:

